August 20th, 2014 – Parkinson’s disease was originally termed Shaking Palsy by James Parkinson when  he became the first to fully characterize the condition in 1817 (1). Since this time the disease has taken on his name despite the fact he never had the condition himself. Parkinson’s disease (PD) is a neurodegenerative condition which outwardly manifests as a movement disorder. PD is chronic, progressive and incurable with an onset usually in the second half of life. Although in a few cases genetic mutations are known to cause PD the specific cause in most cases has not been identified (2).
he became the first to fully characterize the condition in 1817 (1). Since this time the disease has taken on his name despite the fact he never had the condition himself. Parkinson’s disease (PD) is a neurodegenerative condition which outwardly manifests as a movement disorder. PD is chronic, progressive and incurable with an onset usually in the second half of life. Although in a few cases genetic mutations are known to cause PD the specific cause in most cases has not been identified (2).
The list of potential symptoms which have been associated with PD is extensive. The most overt symptoms are related to movement. Tremor of the extremities which appears when they are at rest is often the first symptom to appear. This is accompanied by muscle rigidity and joint stiffness. The most severe symptoms are slowing of movement (bradykinesia) and in extreme cases loss of movement (akinesia). In later stages of PD falls are common due to impaired balance and a general failing of the reflexes which control posture. Stooped shuffling gait and gait freezing are some of the common ways PD effects walking. There are also symptoms of the mouth such as drooling, difficulties swallowing, and changes in, or difficulties with, speech. One suffering from PD may also experience fatigue or an unpleasant desire to move. Neurological symptoms include impaired reaction time and executive functions such as awareness of time, impulse control and awareness/interpretation of interpersonal cues. Memory of how to do things is also often impaired. Insomnia with excessive daytime sleeping and disturbances during REM sleep are also frequently observed. Autonomic functions are also altered producing such things as oily skin, excessive sweating and sexual dysfunction. Motility is also impaired in the gastrointestinal tract resulting in constipation severe enough that it can be life threatening. PD also can impair the eyes reducing blink rate, altering tear production, producing eye twitching and even sometimes producing hallucinations. A disproportionate percentage of those diagnosed with PD also experience depression (58%), apathy (54%), and/or anxiety (49%) (2).
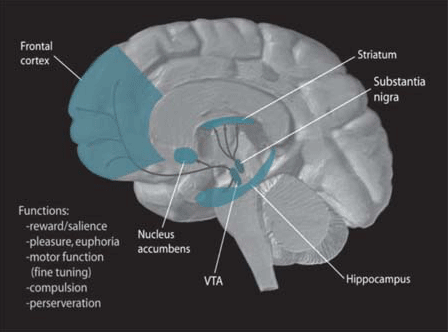
The pathological changes which produce this vast array of symptoms are surprisingly simple. The pigmented dopamine-producing neurons in the pars compacta, a subsection of a relatively small brain structure known as the substantia nigra (latin for “black substance”), start to die off. These neurons send dopamine to the striatum which is a subsection of the basal ganglia. This influx of dopamine plays a role in basal ganglia-produced control of movement and when it starts to dry up two things happen. The direct pathway which facilitates movement is inhibited while the indirect pathway which inhibits movement is facilitated (2). One way to think about it is as if someone stepped on the muscle break while letting up on the muscle throttle. The overall result is things slow down or even stop.
The majority of treatments for PD are designed to address this drop in dopamine production. The most common by far is levodopa (L-dopa) which is the precursor to dopamine. By increase the bioavailability of the dopamine precursor dopamine production increases. Another technique is to inhibit the metabolic breakdown of dopamine with a selective MAO-B inhibitor. If the breakdown of dopamine is inhibited the dopamine which is produced will be more effective for longer. The final pharmacological approach is to activate the dopamine receptors in the striatum and basal ganglia directly with agonists for these receptors. Each of these therapies has their advantages and disadvantages but none of them remain effective indefinitely and the side effects from increased dosages eventually make them intolerable (2).
Drugs which interact with the endocannabinoid system have recently gain attention in the research community as adjunct therapies. Although it is not recommended as a monotherapy for PD today, cannabis was used in the late 1800s as a relative effective treatment for controlling PD associated tremor (1). Today cannabinoids have shown promise as protection against the neurodegeneration of the substantia nigra dopaminergic cells due to their action as powerful antioxidants, potentially to help control tremor, to inhibit the expression of L-dopa induced dyskinesia, to counteract slowing of movement in the case of cannabinoid antagonists and finally as an effective antipsychotic in the case of cannabidiol (CBD).
Parkinson’s-Induced Changes to the Endocannabinoid System
So why might we expect targeting the endocannabinoid system to be of therapeutic value in the treatment of PD? First off, the components of this system such as anandamide, 2-AG, and the cannabinoid receptors are found in high concentration in the basal ganglia and appear to play a role here in the regulation of movement (3). Secondly, movement disorders like PD and even some of the treatments for PD are known to alter the endocannabinoid system in this part of brain. And finally, polymorphisms in CNR1, the gene encoding for the CB1 receptor, have been associated with features of PD.
Changes in the endocannabinoid system have been observed in several of our animal models of PD. In 2000, researchers in the UK observed that naturally occurring anandamide levels in the substantia nigra and the globus pallidus, another part of the basal ganglia affected by PD, were three times higher than had been previously reported elsewhere in the brain. The other primary endocannabinoid, 2-AG, was also found in high concentrations in these two brain regions. They further observed that once Parkinsonism had been induced via reserpine treatment that 2-AG levels in the globus pallidus skyrocketed to seven times what they had been pre-treatment. This elevation of 2-AG was accompanied by the onset of locomotor suppression typical of PD. It is not surprising that selective agonists for either the dopamine D1 or D2 receptor resulted in partial normalization of locomotion after reserpine treatment. After all these receptors are the ultimate final targets for most stimulant drugs. What is perhaps a bit more interesting is that these dopamine agonists resulted in concurrent decrease in both anandamide and 2-AG in the globus pallidus. Furthermore, when the D2 agonist and the CB1 receptor antagonist rimonabant were administered together full normalization of motor activity was restored (4).
The following year the same neuroscientists at the University of Manchester yet again expanded our understanding of the changes to the endocannabinoid system which result from reserpine treatment. This time they observed that CB1 receptor levels in the striatum dropped by 12% medially and 54% in the dorsolateral portion of the striatum following reserpine treatment. They suggested that this might be due to the elevated levels of endocannabinoids previously reported in the basal ganglia after reserpine treatment causing the down regulation of CB1 receptors (5). However, this is not self evident since the elevated basal ganglia endocannabinoid levels which have been found following reserpine treatment were not been found in the striatum.
In another model of PD, 6-hydroxydopamine (6-HO-DA) is administered after which it enters dopaminergic cells were it causes the accumulation of super free radicals which intern kill the dopaminergic cells through severe oxidative stress. When 6-HO-DA is administered to the substantia nigra it produces an experimental Parkinsonism. In 2002, researches in Rome found that this model of PD produced striatal elevations of anandamide but not 2-AG. This elevation appeared to be the result of a generalized decreased anandamide deactivation processes. Activity of both the anandamide reuptake transporter and FAAH, the enzyme responsible for the metabolic deactivation of anandamide, was decreased by 6-HO-DA treatment. This was accompanied by striatal increases in glutamatergic activity which has been associated with muscle rigidity in Parkinsonism. When these Italian scientists administered the super potent synthetic cannabinoid HU-210 similar reductions in glutamatergic activity were seen in the striatum regardless of previous 6-HO-DA exposure. However, when FAAH inhibitors (FAAHIs) were used striatal glutamatergic activity was only substantially reduced if 6-HO-DA exposure had already occurred (6). Together this suggests that the elevation of striatal anandamide observed following 6-HO-DA exposure is a compensatory attempt to correct the over stimulation produced by excessive glutamatergic activity.
Human brain tissue studies have found similar results in the basal ganglia. Compared to the brains of healthy individuals, those who have been diagnosed with PD had reduced CB1 receptor levels in two of the three parts of the striatum and in the globus pallidus (7). Furthermore, the cerebrospinal fluid of PD patients on average contains about twice the amount of anandamide found in healthy controls regardless of past treatment history, stage of PD or its severity (8).
As I mentioned in my last article on depression having two copies of long chain (AAT) alleles in the CNR1 gene appears to offer a statistically significant degree of protection against developing depression in those with PD (9). As mentioned earlier the rate of depression associated with having PD is between three and seven times that seen in the general population (2,10). It is unclear what functional impact this polymorphism in the CNR1 gene has on the endocannabinoid system that might be producing this prophylaxis or how it might impact the expression of other features of PD but these are important avenues of future research into this topic which will further elucidate how manipulating the endocannabinoid system can benefit PD patients.
Cannabinoids as Neuroprotectants against Parkinson’s-Induced Neurodegeneration
There is evidence that cannabinoids play a neuroprotective role in stroke, traumatic brain injury, and several neurodegenerative conditions such as Alzheimer’s disease, Huntington’s disease, Lou Gehrig’s disease, Multiple Sclerosis and even Parkinson’s disease. Cannabinoids appear to produce this protection through various mechanisms. For example, the plant derived cannabinoids like THC and CBD are powerful antioxidants; cannabinoids which activate the CB2 receptor are anti-inflammatory agents by suppressing toxic cytokine release and microglia activation; and cannabinoids which activate the CB1 receptor are anti-excitotoxic due to a suppression of glutamatergic activity, subsequent calcium ion influx and eventual nitric oxide production (11,12). The US Federal Government has such faith in this potential medical benefit of cannabinoids that they have held the US patent on cannabinoids as antioxidants and neuroprotectants since October of 2003. Patent #6,630,507 is assigned to “The United States of America as represented by the Department of Health and Human Services (Washington, DC)” for “Cannabinoids as antioxidants and neuroprotectants” (13).
Several of the neuroprotective properties exerted by cannabinoids may be of particular import to slowing the progression of PD. In the 6-HO-DA model of Parkinsonism, daily THC or CBD was able to significantly attenuate the progression of neurodegeneration which occurred during the first two weeks following exposure to 6-HO-DA. This was not a masking effect either as the benefits of cannabinoid treatment continued even after the two week cannabinoid treatment had come to an end. In cell cultures the super powerful synthetic cannabinoid HU-210 was also able to attenuate 6-HO-DA toxicity although the effect was only substantial when the culture contained both neural and glial cells (14). All three cannabinoids tested are able to activate the CB2 receptor which mediates the antiglial/anti-inflammatory properties of the cannabinoids. In combination with the increased antitoxic effect observed in cell cultures also containing glia over those containing neurons alone, this suggests that immunomodulation produced by CB2 receptor activation may play a primary role in the neuroprotectant properties of cannabinoids. Even so, because the two cannabinoids tested in live animals were both plant derived cannabinoids with well known powerful antioxidant qualities it remains likely that this also played a role in the neuroprotection they produced in the 6-HO-DA model of Parkinsonism.
Another line of evidence which suggests that the endocannabinoid system plays a neuroprotective role in the 6-HO-DA model of Parkinsonism comes from the fact that mice genetically lacking the CNR1 gene which codes for the CB1 receptor, known as CB1 knockout mice, are more vulnerable to the effects of 6-HO-DA toxicity. Not only was the degree of motor impairment greater in the CB1 KO mice compared to their wild type counterparts but they also lost more dopaminergic neurons as well. There was also evidence of increased oxidative and nitric oxide stress in the CB1 KO mice supporting the theory that both antioxidant and antiglial properties play a role in cannabinoid neuroprotection in the 6-HO-DA model of parkinsonism (15). Interestingly this is also the first time we have direct evidence that anti-excitotoxic properties of the endocannabinoid system may be playing a role as well. Something which had only previously been suggested by circumstantial evidence like that fact that cannabinoids inhibit glutamatergic excitotoxicity (11), that excessive striatal glutamatergic activity is inhibited by cannabinoids in the 6-HO-DA model of parkinsonism (6), and glutamatergic/nitric oxide excitotoxicity appears to play a role in the pathogenic destruction of substantia nigra neurons in parkinsonism (16). It is interesting to note that the CB1 KO mice in the above study also did not express L-dopa-induced dyskinesias as severely as was seen by their wild type counterparts (15) and that brings us to our next topic.
Levodopa, dyskinesias and the Endocannabinoid System
One of the primary treatments for PD is the supplementation of dopamine production by the administration of its precursor, L-dopa. That said, systemically administered L-dopa is fraught with substantial drawbacks. For one, only about 1-5% of the administered L-dopa make it into the target neurons to be metabolized into dopamine where needed. The rest is converted to dopamine elsewhere in the body where it creates side effects. Constantly administering systemic L-dopa also activates the natural feedback inhibition controlling L-dopa production and the body stops making its own. This reduces its efficacy requiring more. Eventually, the excessively high doses of L-dopa lead to L-dopa-induced dyskinesias (2). L-dopa-induced dyskinesias are involuntary movements that can appear as jerking, swaying dance-like movement of the upper body (17). This can be almost as disruptive as the disease itself however it does not usually develop except after years of using L-dopa.
L-dopa therapy interestingly appears to alter the endocannabinoid system in the basal ganglia. In the 6-HO-DA model of Parkinsonism neither L-dopa nor 6-HO-DA alone appeared to alter striatal CB1 levels however when 6-HO-DA exposure was followed by chronic L-dopa treatment the expression of striatal CB1 receptors was elevated (18). Through eventual activation of the D1/D2 receptors, L-dopa alone stimulates anandamide release throughout the basal ganglia. On the other hand, 6-HO-DA exposure depletes anandamide in part of the striatum after which L-dopa fails to effect anandamide in the basal ganglia. Furthermore, L-dopa treatment in these animals leads to involuntary dyskinetic movements which are inhibited by administration of the synthetic cannabinoid WIN-55,212-2 (WIN). Together this suggests that an endocannabinoid deficiency plays a role in the expression of L-dopa-induced dyskinesias (19).
Other studies have also shown a beneficial effect of cannabinoids on the expression of L-dopa-induced dyskinesias. Both CB1 receptor agonist (WIN) and the antagonist rimonabant (0.1-3mg/kg) appear to reduce evidence of L-dopa-induced dyskinesia in the reserpine model of Parkinsonism. In this study, anandamide reuptake transporter AM404 did not appear to effect expression of dyskinesia (20). The next study suggests that this lack of effect produced by AM404 might be the result of anandamide’s ability to activation the TRPV1 vanilloid receptor. The dyskinesias produced by L-dopa in the 6-HO-DA model of Parkinsonism appear to be more intense and varied than those produced in the reserpine model. Even so, here again WIN was able to substantially inhibit the expression of dyskinesias. Interestingly the lowest doses tested produced the strongest effect and started working sooner. Again elevating anandamide levels, this time with a FAAHI, had no effect on expression of dyskinesias, that is, unless the FAAHI was co-administered with a TRPV1 antagonist blocking the ability of anandamide to activation this receptor. When this was done anandamide was able to attenuate the expression of dyskinesias more effectively than WIN without any inhibition of the antiparkinsonism produced by the L-dopa(21).
As mentioned above, CB1 receptor antagonists like rimonabant are also able to improve L-dopa-induced dyskinesias. However there appears to be divergence between the two types of dyskinetic inhibition produced by CB1 agonists vs. antagonists such that they reduce different aspects of the dyskinetic expression. Furthermore, they do so most effectively for different points in the L-dopa treatment cycle (22). Antagonists, however, appear to potentially offer another potential benefit as well. They seem to be able to facilitate the effectiveness of L-dopa reducing the dose required to produce therapeutic results. In yet another model of Parkinsonism, using MPTP to induce Parkinsonism, a selective CB1 receptor antagonist had no effect on its own nor were acute does of it able to alter response to L-dopa. However the antiparkinsonism response to threshold doses of L-dopa were potentiated with repeated coadministration of the CB1 antagonist and threshold doses of L-dopa together. This improvement was in the form of a 30% increase in the effective duration of L-dopa-induced antiparkinsonism, such that less L-dopa was required less often to produce the same degree of benefit. The particular CB1 antagonist tested however did not appear to affect the expression of L-dopa-induced dyskinesias in this study (23). These results were collaborated by another study which found that very low doses of the CB1 antagonist rimonabant in the 6-HO-DA model of Parkinsonism were able to produce equal antiparkinsonism effects to a low but effective dose of L-dopa. Furthermore the best results were achieved when both low dose L-dopa and low dose rimonabant (0.05mg/kg) were coadministered. Interestingly, the low dose rimonabant was not effective at ameliorating the expression of dyskinesias produced by larger doses of L-dopa (24). However this might be because the dose used in this study was two to sixty times smaller than that previously associated with inhibition of dyskinesias suggesting different potential antiparkinsonism effects at different doses of rimonabant.
Yet another study has found that rimonabant might be of benefit to PD but only at very specific low doses. In this study, which used a variation of the 6-HO-DA model of Parkinsonism, a 0.1mg/kg dose of rimonabant was able to partially alleviated the reduction and slowing of movement which occurs following 6-HO-DA treatment. Curiously, the improvements in hypokinetic behavior were lost when the dose of rimonabant was increased to 0.5-1mg/kg. The authors were unable to detect any changes in dopaminergic, GABAergic or glutamatergic activity to explain how downstream results of cannabinoid receptor blockade might be producing these improvements (25). Perhaps they were looking in the wrong place, they only checked the striatum. Their results are similar to the case of substantially increased 2-AG in the globus pallidus in the reserpine model of Parkinsonism we discussed earlier under Parkinson’s-Induced Changes to the Endocannabinoid System. Although they are both part of the basal ganglia the globus pallidus lies outside the striatum. This increase in 2-AG in the globus pallidus was associated with hypokinetic behavior which was completely relieved by coadministration of a D2 agonist and a 1mg/kg dose rimonabant (4). Sadly this study did not appear to test rimonabant alone so it we can’t say what dose of rimonabant, if any, might be have been effective alone at alleviated hypokinetic behavior in the reserpine model.
Despite all the positive results of CB1 receptor antagonists like rimonabant in models of parkinsonism the one time it was tested in actual PD patients it produced no discernable antiparkinsonism or antidyskinetic results (26). This could be because only one dose was tested for one acute large dose of L-dopa and the benefits of rimonabant appear to work best in combination with L-dopa with repeat administration (23) or it could be that the dose was too low considering that only one dose was tested and antidyskinetic effects have only been observed at relatively higher doses (20). Then again it is possible that rimonabant simply has no effect on actual PD or L-dopa side effects in humans.
Clinical Trials: Cannabis and other CB1 Agonists
The first study is not a clinical trial as much as a survey taken at a clinic by PD patients who had decided on their own to use cannabis to help treat their PD and reported subjective improvements from the drug. Of the 85 patients who reported using cannabis only one of them took it by inhalation. The other 84 consumed it orally either dry or fresh once a day with meals at an average dose of half a teaspoon. The mean age of these patients was 65.7 and none of them had used cannabis recreationally before learning (usually from the media) that cannabis might be of therapeutic benefit to them. Only four of those responding reported that cannabis actually made their symptoms worse. This survey found that 45.9% experienced general relief, 30.6% relief from tremor, 44.7% relief from slowed/impaired movement, 37.7% relief from rigidity, and 14.1% relief from L-dopa-induced dyskinesias. Except for relief from dyskinesias all of these results were statistically significant. There were some clear effects of length of use and frequency of use such that substantially more of those who had used for three months or more reported relief of symptoms on all measures compared to those who had used cannabis for less than three months. Furthermore, those who used cannabis daily were approximately 3 times more likely to experience relief from dyskinesias than those who used it irregularly. In those who used cannabis regularly for months there also appeared to be an effect of 11-hydroxy-THC on improvements in slowed movement and rigidity. The primary active metabolite of THC especially when consumed orally, 11-HO-THC has both a longer half-life and a stronger effect than THC. When urine content of 11-HO-THC from regularly using patients exceeded 50ng/ml they reported relief from symptoms of slowed/impaired movement and muscle/joint rigidity. If use of cannabis was infrequent or urine content of 11-HO-THC was below 50ng/ml then relief from these two sets of symptoms was not reported. Clearly PD patients are much more likely to experience relief following therapeutic use of cannabis if they use it regularly for more than two months. Frequent use appears to be particularly important for antidyskinetic results (27).
Despite the finding of significant cannabis-produced improvements in PD the survey has one problem, it is subjective and most people in the medical community these days prefer more objective evidence. There is however a clinical study which attempts to address this using a randomized double-blind crossover design. In a 17 patients with PD who experienced dyskinesias from L-dopa therapy no effect on expression of dyskinesias was observed following daily oral cannabis extract. Indeed no reliable pro or anti-parkinsonism effects produced by cannabis were observed at all (28). On issue with this particular study which is suggested by the aforementioned survey is that perhaps the treatment periods did not last long enough since on average at least 1.7 months were required before patients experienced noticeable relief and the treatment periods in this study only lasted one month. Or perhaps the dose was not large enough. Another issue concerns relative concentrations of THC and CBD in the extract. As with anandamide, CBD activates the TRPV1 receptor (29) and therefore if in too high of a relative concentration in the extract it may be masking the antidyskinetic effects produced by the THC at the CB1 receptors (21).
By far the most promising trial on the use of cannabinoids to alleviate L-dopa induced dyskinesias comes from a randomized, double-blind, placebo-controlled, crossover pilot study on seven PD patients using nabilone. Nabilone, aka Cesamet, is a synthetic cannabinoid structurally derived from THC and with a similar overall potency and pharmacology to THC. In this study nabilone was found to effectively reduce L-dopa-induced dyskinesias but appeared to be most effective against dyskinesia produced by diphasic vs. peak dose and for dystonia vs. chorea-type dyskinesias. During L-dopa-induced dyskinesia the lateral globus pallidus appears to be hyperactive and activating the CB1 receptors in this region increases GABAergic tone. The role of GABA is to slow things down by reducing the firing rate of the cells to which it binds. The authors therefore suggested that the nabilone was producing its antidyskinetic effect by binding to the CB1 receptors in the lateral globus pallidus thereby increasing GABAergic tone and inhibiting the hyperactivity of this brain region resulting in less symptoms of dystonia (30).
There is one final clinical trial in the use of cannabinoids to aid in the treatment of PD patients which bares mention here. In my article in issue #20 Cannabis and Melatonin as Mood Regulators for a Woman of 38 with Unipolar, Rapid-Cycling Mania I discuss a double case report on the use of CBD as an antipsychotic in two women diagnosed with bipolar disorder which found CBD was ineffective at reducing psychotic symptoms in these two women (31). So far the evidence appears to be more positive for CBD when it comes to controlling the expression of psychotic symptoms in PD patients. The same researchers studying the two manic women conducted a pilot study on six PD patients who had experienced symptoms of psychosis for at least three months. They found that in these four men and two women that not only was CBD well tolerated over the four week trial but that by the end CBD had lowered total scores on the Unified Parkinson’s Disease Rating Scale as well as significantly reducing expression of psychotic symptoms (32). This finding is important not only because it demonstrates that CBD might possess mild antiparkinsonism properties on its own but because it can safely and significantly reduce psychosis symptoms in PD patients. This is something which has proven challenging in the past since most historically used antipsychotics inhibit the dopaminergic system and thereby negatively impacting Parkinsonism.
Conclusion
Parkinson’s disease is a chronic, presently incurable neurodegenerative condition resulting in a movement disorder. It is primarily caused by the loss of substantia nigra dopaminergic neurons which deregulates the downstream control of movement in the rest of the basal ganglia, particularly the striatum and globus pallidus. This results in slowed/impaired movement or even the cessation of movement, tremor, and rigidity. The primary drug used to alleviate the symptoms, levodopa, eventually requires such large doses that it produces involuntary movements as a side effect which can be almost as disruptive as the disease itself.
Downstream alterations to the endocannabinoid system throughout the basal ganglia are among the many changes which result from the loss of the dopaminergic neurons in the substantia nigra. These alterations include changes in CB1 receptor densities as well as in the production of both anandamide and 2-AG, two of the primary endocannabinoids. Administration of L-dopa in cases of Parkinsonism is also able to alter the endocannabinoid system in the basal ganglia. However the lack of D2 receptor activation in the striatum produced by the disease itself results in an endocannabinoid deficiency preventing the increase in striatal anandamide normally following L-dopa administration in the healthy brain. Some of these changes appear to be compensatory in nature and are the body’s natural attempt to combat the loss of stimulation from the substantia nigra. Others, especially those produced by L-dopa are more pathogenic in nature and may serve to facilitate the expression of slowed movement and dyskinesias.
One of the potential therapeutic applications of cannabinoids is to slow the progression of the disease through the numerous neuroprotective properties they possess but especially their antioxidant, anti-excitotoxic, and antiglial/anti-inflammatory properties. This would likely be more effective when applied early on in the disease progression and therefore might be able to be used most effectively in known cases of genetic mutation produced PD because CBD therapy for example could start before evidence of the disease ever developed. Other evidence suggests that cannabinoids might be effective at relieving tremor, slowing of movement, and rigidity while ameliorating the dyskinetic side effects of L-dopa. Somewhat confusingly, CB1 receptor antagonists like rimonabant have also been shown to be effective against the slowing of movement in models of Parkinsonism as well as for reducing L-dopa-induced dyskinesias in these models. However the first test of CB1 antagonists in humans was not successful. There are several possible reasons why this test failed and it is quite possible that a more thorough trial might yet find a positive result of the use of CB1 antagonists in the treatment of PD in humans. Finally in a pilot study, CBD was found to produce significant antipsychotic effects in 6 cases of PD with psychosis, a condition which historically has been especially challenging to treat since typical antipsychotics can exacerbate PD symptoms.
There is evidence that for those wishing to try and treat themselves with cannabis that it may be more effective at alleviating the symptoms of the disease itself. However if relief from L-dopa induced dyskinesia is the goal then it might be best to avoid strains high in CBD since it is known to active the TRPV1 receptor and this can mask the antidyskinetic effect produced by activating the CB1 receptors with THC. So far once daily oral doses in the range of 0.25-0.5g for more than two months appears to be required to start to experience quality results. Although higher doses might be require for some, this dose range appears adequate for producing results with minimal side effects. Surprisingly, despite the fact that cannabis can induce sleep and problems sleeping are frequently associated with PD there has been no apparent attempt in the literature to investigate this possible beneficial impact from oral cannabis use in PD patients. Although also not addressed in the literature, one potential negative impact from using cannabis with PD is that it could exacerbate the PD-associated impaired GI motility since it too slows GI tract motility. Therefore it might be best to skip cannabis treatments if one is experiencing significant constipation.
Drugs which increase anandamide tone through the blockade of reuptake and/or its metabolic breakdown are also likely to be beneficial although would need to be coadministered with a TRPV1 antagonist if harnessing the antidyskinetic properties of anandamide was desired. On the other hand, decreasing 2-AG tone may ameliorate some of the slowing of movement produced by PD. It is likely that drugs which directly affect the endocannabinoid system in these ways, such as AM404 and the FAAHIs, are the future of mainstream cannabinoid based treatments for Parkinson’s disease.
References
1. Gowers, WR. A manual of diseases of the nervous system. London; v. II, 1886, 589-607.
2. Wikipedia. Parkinson’s disease. http://en.wikipedia.org/wiki/Parkinson%27s_disease (Accessed 1-27-2010).
3. Fernández-Ruiz, J. The endocannabinoid system as a target for the treatment of motor dysfunction. British Journal of Pharmacology, 2009 Apr; 156 (7): 1029-40.
4. Di Marzo, V, Hill, MP, Bisogno, T, Crossman, AR, and Brotchie, JM. Enhanced levels of endogenous cannabinoids in the globus pallidus are associated with a reduction in movement in an animal model of Parkinson’s disease. FASEB Journal, 2000 Jul; 14 (10): 1432-8.
5. Silverdale, MA, McGuire, S, McInnes, A, Crossman, AR, and Brotchie, JM. Striatal cannabinoid CB1 receptor mRNA expression is decreased in the reserpine-treated rat model of Parkinson’s disease. Experimental Neurology, 2001 Jun; 169 (2): 400-6.
6. Gubellini, P, Picconi, B, Bari, M, Battista, N, Calabresi, P, Centonze, D, Bernardi, G, Finazzi-Agrò, A, and Maccarrone, M. Experimental parkinsonism alters endocannabinoid degradation: implications for striatal glutamatergic transmission. Journal of Neuroscience, 2002 Aug 15; 22 (16): 6900-7.
7. Hurley, MJ, Mash, DC and Jenner, P. Expression of cannabinoid CB1 receptor mRNA in basal ganglia of normal and parkinsonian human brain. Journal of Neural Transmission, 2003 Nov; 110 (11): 1279-88.
8. Pisani, A, Fezza, F, Galati, S, Battista, N, Napolitano, S, Finazzi-Agrò, A, Bernardi, G, Brusa, L, Pierantozzi, M, Stanzione, P, and Maccarrone, M. High endogenous cannabinoid levels in the cerebrospinal fluid of untreated Parkinson’s disease patients. Annals of Neurology, 2005 May; 57 (5): 777-9.
9. Barrero, FJ, Ampuero, I, Morales, B, Vives, F, de Dios Luna Del Castillo, J, Hoenicka, J, and García Yébenes, J. Depression in Parkinson’s disease is related to a genetic polymorphism of the cannabinoid receptor gene (CNR1). Pharmacogenomics Journal, 2005; 5 (2): 135-41.
10. Wikipedia. Major Depression: Epidemiology. http://en.wikipedia.org/wiki/Major_depression#Epidemiology (Accessed 1-28-2010).
11. Martínez-Orgado, J, Fernández-López, D, Lizasoain, I and Romero, J. The seek of neuroprotection: introducing cannabinoids. Recent Patents on CNS Drug Discovery, 2007 Jun; 2 (2): 131-9.
12. Romero, J and Orgado, JM. Cannabinoids and neurodegenerative diseases. CNS & Neurological Disorders Drug Targets, 2009 Dec; 8 (6): 440-50.
13. Hampson, AJ, Axilrod, J and Grimaldi, M. Cannabinoids as antioxidants and neuroprotectants. http://patft.uspto.gov/netahtml/PTO/srchnum.htm (Patent #6,630,507 Accessed 1-29-2010).
14. Lastres-Becker, I, Molina-Holgado, F, Ramos, JA, Mechoulam, R, and Fernández-Ruiz, J. Cannabinoids provide neuroprotection against 6-hydroxydopamine toxicity in vivo and in vitro: relevance to Parkinson’s disease. Neurobiology of Disease, 2005 Jun-Jul; 19 (1-2): 96-107.
15. Pérez-Rial, S, García-Gutiérrez, MS, Molina, JA, Pérez-Nievas, BG, Ledent, C, Leiva, C, Leza, JC, and Manzanares, J. Increased vulnerability to 6-hydroxydopamine lesion and reduced development of dyskinesias in mice lacking CB1 cannabinoid receptors. Neurobiology of Aging, 2009 May 4. [Epublished ahead of print].
16. Beal, MF. Excitotoxicity and nitric oxide in Parkinson’s disease pathogenesis. Annals of Neurology, 1998 Sep; 44 (3 Suppl 1): S110-4.
17. Wikipedia. Dyskinesia. http://en.wikipedia.org/wiki/Dyskinesia (Accessed 1-29-2010).
18. Zeng, BY, Dass, B, Owen, A, Rose, S, Cannizzaro, C, Tel, BC, and Jenner, P. Chronic L-DOPA treatment increases striatal cannabinoid CB1 receptor mRNA expression in 6-hydroxydopamine-lesioned rats. Neuroscience Letters, 1999 Dec 3; 276 (2): 71-4.
19. Ferrer, B, Asbrock, N, Kathuria, S, Piomelli, D, and Giuffrida, A. Effects of levodopa on endocannabinoid levels in rat basal ganglia: implications for the treatment of levodopa-induced dyskinesias. European Journal of Neuroscience, 2003 Sep; 18 (6): 1607-14.
20. Segovia, G, Mora, F, Crossman, AR and Brotchie, JM. Effects of CB1 cannabinoid receptor modulating compounds on the hyperkinesia induced by high-dose levodopa in the reserpine-treated rat model of Parkinson’s disease. Movement Disorders, 2003 Feb; 18 (2): 138-49.
21. Morgese, MG, Cassano, T, Cuomo, V and Giuffrida, A. Anti-dyskinetic effects of cannabinoids in a rat model of Parkinson’s disease: role of CB(1) and TRPV1 receptors. Experimental Neurolory, 2007 Nov; 208 (1): 110-9.
22. Fabbrini, G, Brotchie, JM, Grandas, F, Nomoto, M and Goetz, CG. Levodopa-induced dyskinesias. Movement Disorders, 2007 Jul 30; 22 (10): 1379-89.
23. Cao, X, Liang, L, Hadcock, JR, Iredale, PA, Griffith, DA, Menniti, FS, Factor, S, Greenamyre, JT, and Papa, SM. Blockade of cannabinoid type 1 receptors augments the antiparkinsonian action of levodopa without affecting dyskinesias in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated rhesus monkeys. Journal of Pharmacology and Experimental Therapeutics, 2007 Oct; 323 (1): 318-26.
24. Kelsey, JE, Harris, O and Cassin, J. The CB(1) antagonist rimonabant is adjunctively therapeutic as well as monotherapeutic in an animal model of Parkinson’s disease. Behavioral Brain Research, 2009 Nov 5; 203 (2): 304-7.
25. González, S, Scorticati, C, García-Arencibia, M, de Miguel, R, Ramos, JA, and Fernández-Ruiz, J. Effects of rimonabant, a selective cannabinoid CB1 receptor antagonist, in a rat model of Parkinson’s disease. Brain Research, 2006 Feb 16; 1073-1074: 209-19
26. Mesnage, V, Houeto, JL, Bonnet, AM, Clavie,r I, Arnulf, I, Cattelin, F, Le Fur, G, Damier, P, Welter, ML, and Agid, Y. Neurokinin B, neurotensin, and cannabinoid receptor antagonists and Parkinson disease. Clinical Neuropharmacology, 2004 May-Jun; 27 (3): 108-10.
27. Venderová, K, R?zicka, E, Vorísek, V and Visnovský, P. Survey on cannabis use in Parkinson’s disease: subjective improvement of motor symptoms. Movement Disorders, 2004 Sep; 19 (9): 1102-6.
28. Carroll, CB, Bain, PG, Teare, L, Liu, X, Joint, C, Wroath, C, Parkin, SG, Fox, P, Wright, D, Hobart, J, and Zajicek, JP. Cannabis for dyskinesia in Parkinson disease: a randomized double-blind crossover study. Neurology, 2004 Oct 12; 63 (7): 1245-50.
29. Bisogno, T, Hanus, L, De Petrocellis, L, Tchilibon, S, Ponde, DE, Brandi, I, Moriello, AS, Davis, JB, Mechoulam, R, and Di Marzo, V. Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. British Journal of Pharmacology, 2001 Oct; 134 (4): 845-52.
30. Sieradzan, KA, Fox, SH, Hill, M, Dick, JP, Crossman, AR, and Brotchie, JM. Cannabinoids reduce levodopa-induced dyskinesia in Parkinson’s disease: a pilot study. Neurology, 2001 Dec 11; 57 (11): 2108-11.
31. Zuardi, A, Crippa, J, Dursun, S, Morais, S, Vilela, J, Sanches, R, and Hallak, J. Cannabidiol was ineffective for manic episode of bipolar affective disorder. Journal Psychopharmacology, 2008 Nov 21 [Epublished ahead of print].
32. Zuardi, AW, Crippa, JA, Hallak, JE, Pinto, JP, Chagas, MH, Rodrigues, GG, Dursun, SM, and Tumas, V. Cannabidiol for the treatment of psychosis in Parkinson’s disease. Journal of Psychopharmacology, 2009 Nov; 23 (8): 979-83.<-->